
Et si l’outil principal des chimistes depuis 30 ans était sur le point d’être dépassé ?
La chimie numérique, aussi appelée chimie computationnelle, consiste à utiliser des modèles physiques et des logiciels pour prédire le comportement des molécules, leur structure, leur énergie ou encore leur réactivité, sans avoir à réaliser chaque expérience dans la réalité.
Concrètement, ces outils permettent de simuler la forme d’une molécule, la manière dont elle réagit avec une autre, les propriétés d’un matériau ou encore la vitesse d’une réaction chimique.
Tout cela repose sur un compromis permanent : plus on veut de précision, plus le calcul devient lourd.
C’est précisément ce verrou que les chercheurs de l’ETH Zurich cherchent à faire sauter avec une nouvelle génération de modèles.
Les potentiels interatomiques d’apprentissage automatique (en anglais machine learning interatomic potentials ou MLIPs) pourraient remplacer la théorie de la fonctionnelle de la densité (DFT) dans la prochaine décennie.
Cela a l’air très technique mais nous allons vous expliquer pourquoi cela pourrait accélérer la découverte de nouveaux matériaux, de médicaments et de procédés industriels à une vitesse inédite !
Lire aussi :
- La France et le Japon dévoilent la plus grosse révolution en cryptographie depuis 90 ans et la machine de Turing avec l’ADN comme support
- La France abrite la 4e plus grande réserve de corail au monde et une technique électrique américaine pourrait permettre de gagner assez de temps pour la sauver
Un nouveau paradigme pourrait bouleverser la chimie numérique
La chimie moderne repose sur une carte invisible
Pour comprendre l’enjeu, imaginez que chaque molécule possède une sorte de relief énergétique invisible.
Les chimistes appellent cela une surface d’énergie potentielle (SEP).
Cette surface indique :
- quelles structures sont stables
- comment une réaction chimique peut se produire
- à quelle vitesse elle se déroule
Depuis des décennies, la Théorie de la Fonctionnelle de la Densité (DFT) est l’outil principal pour calculer cette carte.
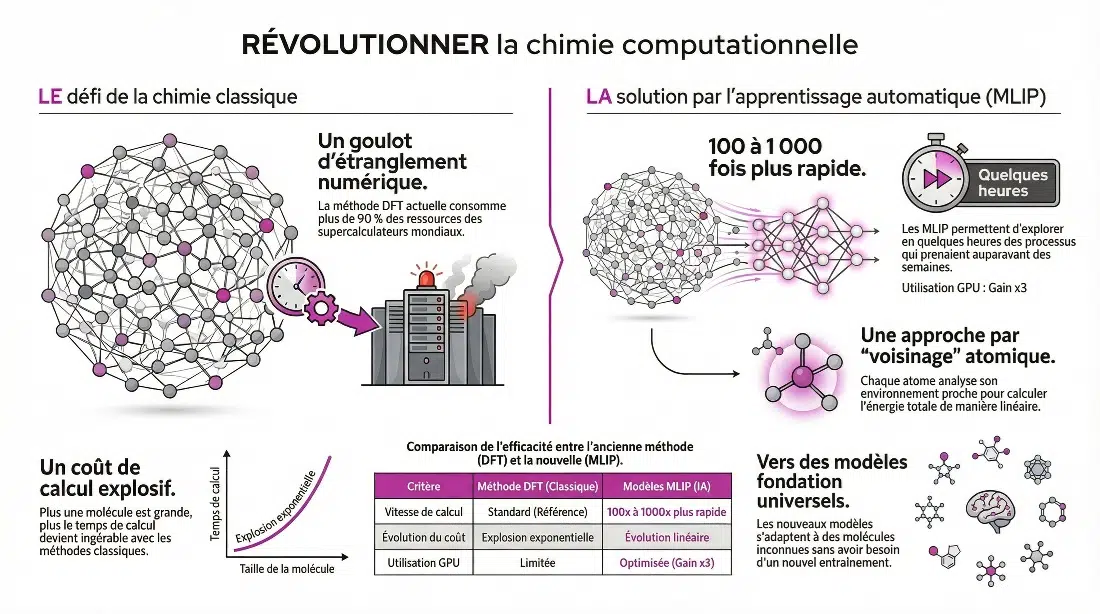
Elle représente aujourd’hui plus de 90 % des calculs de chimie quantique et consomme une part significative des supercalculateurs mondiaux.
Le problème, c’est qu’elle reste coûteuse en calcul et repose sur des approximations difficiles à améliorer systématiquement.
Les MLIPs : la vitesse des modèles classiques, la précision du quantique
Les MLIPs changent complètement la manière d’aborder le problème.
Au lieu de résoudre directement les équations de la mécanique quantique à chaque étape, ces modèles utilisent l’intelligence artificielle pour apprendre le comportement des atomes.
Le principe est simple. Un système chimique complexe est découpé en contributions locales :
- chaque atome « regarde » ses voisins proches
- il calcule son énergie locale
- l’énergie totale est la somme de toutes ces contributions
De cette manière le coût de calcul évolue de manière linéaire avec le nombre d’atomes, au lieu d’exploser comme dans les méthodes classiques.
Des gains de performance spectaculaires
Après tests, les scientifiques de l’ETH Zurich affirment que les MLIPs sont :
- 100 à 1 000 fois plus rapides que les calculs DFT pour certaines optimisations
- capables de simuler des systèmes de milliers d’atomes
- exploitables efficacement sur GPU, avec un gain moyen d’environ ×3
Cette accélération permet de simuler des phénomènes sur des échelles de temps beaucoup plus longues.
Des processus qui prenaient des semaines peuvent être explorés en quelques heures.
Pour la recherche industrielle, cela ouvre des perspectives concrètes comme la conception accélérée de catalyseurs, la découverte de nouveaux matériaux ou l’optimisation de batteries.
Des modèles capables de généraliser au-delà des cas étudiés
Historiquement, les modèles de ce type étaient spécifiques à un système donné.
Il fallait entraîner un modèle pour chaque molécule ou matériau étudié.
Aujourd’hui, une nouvelle génération apparaît : les modèles fondation.
Ces modèles sont entraînés sur des bases de données gigantesques, parfois des centaines de millions de structures chimiques.
Ils deviennent capables de fonctionner sur des systèmes qu’ils n’ont jamais vus.
Deux approches coexistent :
- modèle généraliste : utilisé tel quel pour explorer rapidement de nombreux systèmes
- modèle affiné : ajusté sur un cas précis pour atteindre une précision maximale
Cette flexibilité permet d’adapter l’outil au besoin, entre rapidité et précision.
Une nouvelle façon de comprendre les molécules
Les MLIPs modernes utilisent des architectures inspirées des réseaux neuronaux graphiques.
Concrètement :
- les atomes sont des nœuds
- leurs interactions sont des liens
- les informations circulent entre eux
À chaque étape, chaque atome met à jour sa compréhension de son environnement.
Ce mécanisme, appelé passage de message, permet de capturer des interactions complexes entre plusieurs atomes.
Les modèles les plus avancés conservent même l’information directionnelle, ce qui est essentiel pour calculer :
- les forces
- les dipôles
- les propriétés optiques
On ne se contente plus de calculer une énergie, on peut désormais reconstruire une image dynamique du système !
Les limites actuelles : tout n’est pas encore réglé
Malgré leurs performances, ces modèles doivent encore franchir plusieurs obstacles.
Les chercheurs identifient notamment :
- la gestion des interactions à longue distance, comme les forces électrostatiques
- la prise en compte de la charge électrique et du spin (manière dont l’état d’une particule évolue lorsqu’on la fait pivoter dans l’espace), essentiels en chimie réelle
- le calcul efficace des dérivées secondes (utiles pour les spectres et les états de transition)
- la quantification de l’incertitude, indispensable pour savoir si un résultat est fiable
Ces défis ne bloquent pas l’utilisation des MLIPs mais ils définissent les prochaines étapes de leur développement.
Un changement de paradigme déjà en marche
Ce qui ressort de cette étude, ce n’est pas seulement une amélioration technique.
C’est un véritable changement de rôle des outils.
Dans le futur proche :
- les MLIPs fourniront la première approximation rapide
- les méthodes quantiques classiques serviront de référence pour valider et affiner
La chimie numérique entre donc dans une phase où la vitesse ne se fait plus au détriment de la précision.
Comprendre les potentiels interatomiques d’apprentissage automatique (MLIPs) en un coup d’oeil :
Source :
Husistein, R. T., & Reiher, M. (2026). A New Paradigm for Computational Chemistry. arXiv.
https://doi.org/10.48550/arXiv.2604.01360 [Preprint]
Image de mise en avant : Représentation 3D de l’ADN (Freepik)

{kind=link}